Histopathology
DECALCIFICATION
Introduction:- The presence of calcium salts in tissues makes them hard. This causes damage to the knife, difficulty in cutting tissue. Calcium is normally present in bones and teeth. Calcium may also be present in normal tissues in pathological conditions like necrotic tissue in tuberculosis. OBJECTIVES:- After reading this lesson, you will be able to: describe decalcification explain different methods of decalcification describe the chemical and physical tests to estimate the remaining calcium DECALCIFICATION:-
FIXATION OF TISSUES
Introduction:- It is a process by which the cells or tissues are fixed in chemical and partly physical state so that they can withstand subsequent treatment with various reagents, with minimal distortion of morphology and no decomposition OBJECTIVES:- After reading this lesson, you will be able to: *state the aims of fixation *explain the principle of fixation *describe the properties and factors affecting fixation *explain types of fixation. AIMS OF FIXATION:- (a) To preserve the tissues as close to their living state as possible (b) To prevent autolysis and bacterial attack (c) To prevent tissues from changing their shape and size during processing (d) To harden the tissues (e) To allow clear staining of sections subsequently (f) To improve the optical differentiation of cells & tissue. PRINCIPLE OF FIXATION:- Fixation results in denaturation and coagulation of protein in the tissues. The fixatives have a property of forming cross links between proteins, thereby forming a gel, keeping everything in their in vivo relation to each other. PROPERTIES OF FIXATIVES AND FACTORS AFFECTING FIXATION:-1. Coagulation and precipitation of proteins in tissues. 2. Penetration rate differs with different fixatives depending on the molecular weight of the fixative 3. pH of fixatives – Satisfactory fixation occurs between pH 6 and 8. Outside this range, alteration in structure of cell may take place. 4. Temperature – Room temperature is alright for fixation. At high temperature there may be distortion of tissues. 5. Volume changes – Cell volume changes because of the membrane permeability and inhibition of respiration. 6. An ideal fixative should be cheap, nontoxic and non-inflammable. The tissues may be kept in the fixative for a long time.TYPE OF FIXATION:-*Immersion fixation *Perfusion fixation *Vapour fixation *Coating/Spray fixation *Freeze drying *Microwave fixation/StabilizationThe most commonly used technique is simple immersion of tissues/smears in an excess of fixative. For all practical purposes immersion fixatives are most useful. These may be divided into routine and special. SIMPLE FIXATIVES:-1. Formaldehyde: Commercially available solution contains 35%-40% gas by weight, called as formalin. Formaldehyde is commonly used as 4% solution, giving 10% formalin for tissue fixation. Formalin is most commonly used fixative. It is cheap, penetrates rapidly and does not over- harden the tissues. The primary action of formalin is to form additive compounds with proteins without precipitation. Formalin brings about fixation by converting the free amine groups to methylene derivatives If formalin is kept standing for a long time, a large amount of formic acid is formed due to oxidation of formaldehyde and this tends to form artefact which is seen as brown pigment in the tissues. To avoid this buffered formalin is used. 2. Absolute alcohol – it may be used as a fixative as it coagulates protein. Due to its dehydrating property it removes water too fast from the tissues and produces shrinkage of cells and distortion of morphology. It penetrates slowly and over-hardens the tissues.3. Acetone:-Sometimes it is used for the study of enzymes especially phosphatases and lipases. Disadvantages are the same as of alcohol. 4. Mercuric chloride:- It is a protein precipitant. However it causes great shrinkage of tissues hence seldom used alone. It gives brown colour to the tissues which needs to be removed by treatment with Iodine during dehydration. 5. Potassium dichromate:- It has a binding effect on protein similar to that of formalin. Following fixation with Potassium dichromate tissue must be well washed in running water before dehydration. 6. Osmic acid :- It is used for fixation of fatty tissues and nerves.7. Chromic acid:- It precipitates all proteins and preserves carbohydrates. Tissues fixed in chromic acid also require thorough washing with water before dehydration. 8. Osmium tetraoxide:- It gives excellent preservation of cellular details, hence used for electron-microscopy. 9. Picric acid – It precipitates proteins and combines with them to form picrates. Owing to its explosive nature when dry; it must be kept under a layer of water. Tissue fixed in picric acid also require thorough washing with water to remove colour. Tissue can not be kept in picric acid more than 24 hrs. COMPOUND FIXATIVES:- 1. Formal saline:-It is most widely used fixative. Tissue can be left in this for long period without excessive hardening or damage. Tissues fixed for a long time occasionally contain a pigment (formalin pigment). This may be removed in sections before staining by treatment with picric alcohol or 10% alcoholic solution of sodium hydroxide. The formation of this pigment can be prevented by neutralizing or buffering the formal saline.Fixation time – 24 hours at room temprature2. Formal calcium:-Useful for demonstration of phospholipids. Fixation time-24 hours at room temperature3. Zenker’s fluid:-It contains mercuric chloride, potassium-di-chromate, sodium sulphate and glacial acetic acid.Advantages :-even penetration, rapid fixationDisadvantages –After fixation the tissue must be washed in running water to remove excess dichromate. Mercury pigment must be removed with Lugol’s iodine.4. Zenker’s formal (Helly’s fluid):-In stock Zenker’s fluid, formalin is added instead of acetic acid.Advantages :- excellent microanatomical fixative especially for bone marrow, spleen & kidney.5. Bouins fluid :-It contains picric acid, glacial acetic acid and 40% formaldehyde.Advantages :-(a) Rapid and even penetration
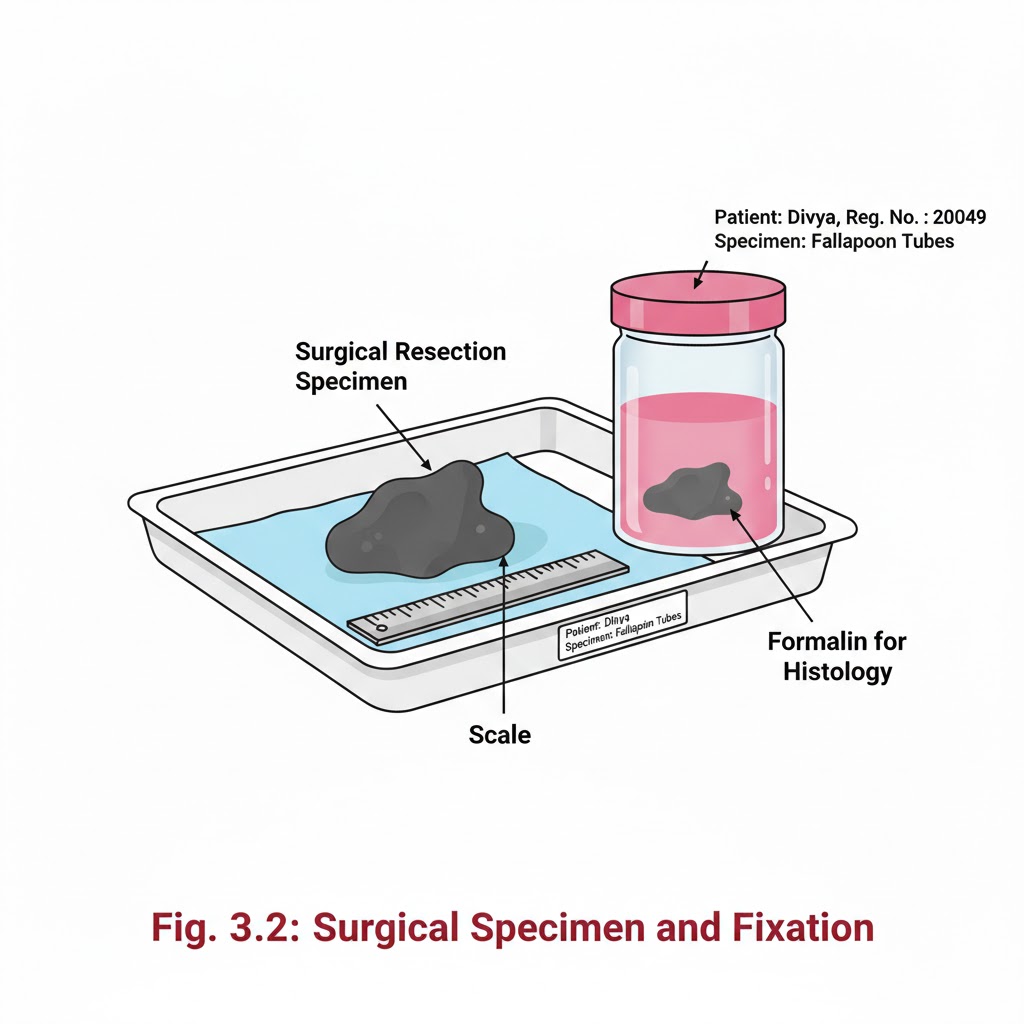
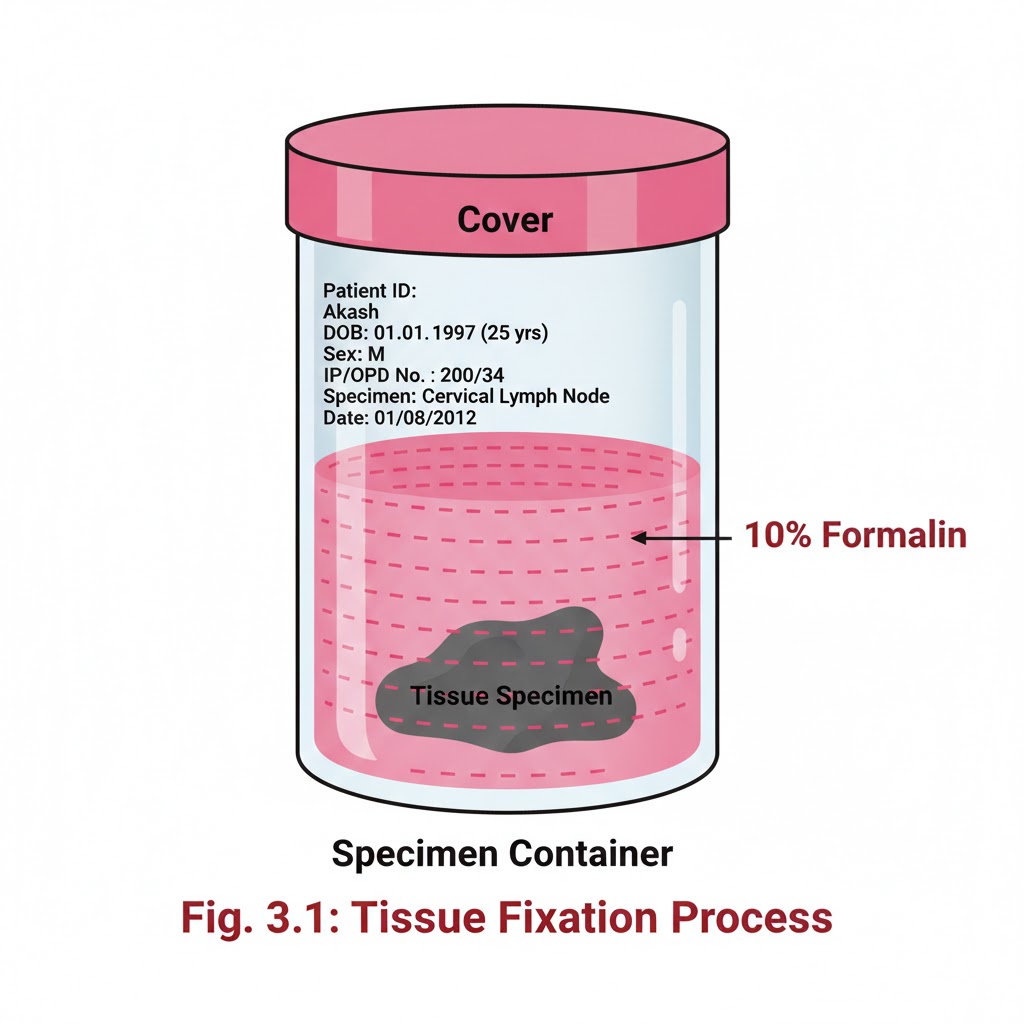
RECEIVING OF SURGICAL SPECIMENS
INTRODUCTION:- In the laboratory setting, numerous histological specimens are received throughout the day for testing. Receiving of Surgical Specimens requires a systematic approach to ensure that all samples are properly accounted for and handled correctly. Without proper documentation and verification, there is a potential to misplace or lose samples. OBJECTIVES:-After reading this lesson, you will be able to:- *explain the process of receiving surgical specimens *describe preparation of gross room *receive the samples, label the sample and store. RECEIVING OF SURGICAL SPECIMENS:- At the time of receiving the specimens, following points should be checked and these points must match between requisition form and label on the sample container.1. Name of the patient 2. Sex and age of patient 3. Registration no, OPD or indoor number 4. Type of sample like appendix or lymph node. After matching the above points carefully, accession number of the Histopathology laboratory should be given on the requisition form and on the sample container like it has been depicted in the form and sample bottle. A register should be maintained for record and for future reference Following points should be noted on the register (sample given) 1.Date 2.Accession number which was given by the pathology department. 3.Patients name, age, sex. 4.Patients registration number/ OPD/ indoor number. 5.Type of sample. 6.Number of samples received from one patient. 7.Remarks / final diagnosis which may be entered later on. After receiving the samples the consultant should be informed for grossing. If grossing to be done after some time, fixative should be put in all the samples to prevent autolysis of the specimen. PREPARATION OF GROSS ROOM:- The routine work associated with a surgical pathology specimen includes gross and microscopic examination. Proper preservation of tissues and processing of the tissue are the most important aspects for correct diagnosis. The size and features of the gross room depends on the number of specimens and type of institution. Gross room should be well illuminated and ventilated.It should have a gross station and racks to keep the specimen in order of accession number. Gross station – It should have ventilated hood. 1. Cutting board placed inside the metal box designed in such a fashion that all the fluids should flow directly into the sink 2. Ready access to sink with hot & cold water 3. Formalin – stock and 10% buffered formalin 4. Box of instruments containing (a) Scissors (b) Forceps (c) Malleable probe (d) Scalpel handle with disposable blades (e) Long knife (f) Scale (g) Pins for attaching the specimens to corked surface if required 5. Containers with different fixatives 6. Bone cutter 7. Large disposal bin 8. Box with cassettes and labels These are the essential items. Depending upon the pathology service being rendered to the institution more sophisticated items may be added. Apart from the above items following items may be of help in keeping the records 1. Photographic facility 2. Refrigerator 3. Balance to weigh the gross specimen 4. X-ray view box 5. Other equipments for tissue bank facility.
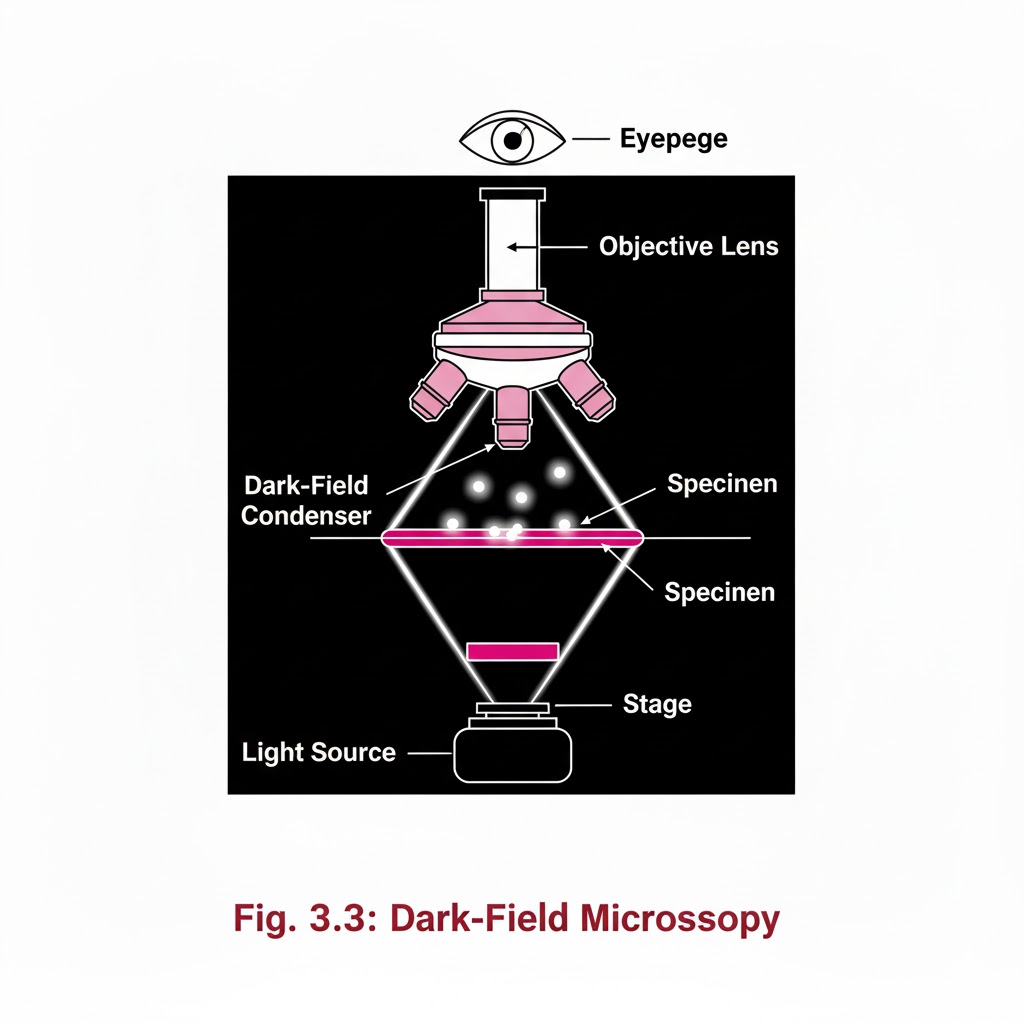
special light microscope
Introduction:- Special Light Microscope refers to advanced microscopic techniques used to examine specimens that cannot be clearly observed under a simple compound microscope. Microscopes are instruments designed to produce magnified visual or photographic images of objects too small to be seen with the naked eye. A microscope must accomplish three tasks: produce a magnified image of the specimen, separate the details in the image, and render those details visible to the human eye or camera. Compound microscopes are suitable for examination of stained preparations, but for special conditions we require a Special Light Microscope such as dark-ground, phase contrast, polarizing, and immunofluorescence microscopes. Objectives of Special Light Microscope After reading this lesson, you will be able to: *describe the principle of Dark-ground, phase contrast, polarizing and immunofluorescence microscopy. *explain the uses of Dark-ground, phase contrast, polarizing and immunofluorescence microscopy. DARK GROUND ILLUMINATION:- Conditions arise when specimen need to be visualized as unstained or living cells. Since such specimens have refractive indices close to medium in which they are suspended, bright field microscopy is difficult as there is not much contrast. Principle:- Dark ground microscopy prevents direct light from entering the front of the objective, only light which enters is which gets reflected or diffracted by the specimen, thus making them appear bright in a dark background . Uses of Dark ground microscopy::- Useful for spirochaetes, flagellates, cell suspensions, flow cell techniques, parasites, autoradiography, and fluorescence microscopy. Disadvantage: Resolution is inferior to bright field microscopy. Does not reveal internal details PHASE CONTRAST MICROSCOPY:- Unstained and living biological material viewing by bright field and dark ground illumination has problems of reduced illumination and resolution. To overcome these problems, phase contrast microscopes are used. Principle: It is an optical microscopy illumination technique that converts phase shifts in light passing through a transparent specimen to brightness changes in the image. The phase shifts themselves are invisible to the human eye, but become visible when they are shown as brightness changes. A practical implementation of phase-contrast illumination consists of a phase ring (located in an aperture plane located somewhere behind the front lens element of the objective) and a matching annular ring, which is located in the conjugate primary aperture plane Two selected light rays, which are emitted from one point inside the lamp’s filament, are focused by the field lens exactly inside the opening of the condenser annular ring. Since this location is precisely in the front focal plane of the condenser, the two light rays are then refracted in such way that they exit the condenser as parallel rays. Assuming that the two rays in question are neither refracted nor diffracted in the specimen plane (location of microscope slide), they enter the objective as parallel rays. Since all parallel rays are focused in the back focal plane of the objective, the back focal plane is a conjugate aperture plane to the condenser’s front focal plane (also location of the condenser annulus). To complete the phase setup, a phase plate is positioned inside the back focal plane in annulus Uses: It’s a quick and efficient way of examining unstained paraffin, resin and frozen sections, studying living cells (cell cultures) and their behavior. POLARIZED LIGHT MICROSCOPY:- Light can be described as an electromagnetic vibration where there are many planes of vibration. Natural light vibrates in many planes or directions, whereas polarized light vibrates in only one plane. It can be produced by passing light through a polarizer. Substances capable of producing polarized light are called birefringent. Principle: The dedicated polarizing microscope uses two polarizers. One, always referred to as polarizer, is placed beneath the substage condenser. The other is called analyzer and is placed between the objective and the eyepiece. Looking through both polarizers, the light intensity is best when they are both giving light vibrating parallel to each other. However, when the light vibration is at right angles to each other, there is dark background. If a substance capable of birefringence is placed between the two polarisers it gets visualized as brightness against a black background. Uses: Medicine-Amyloid detection, collagen fibers, urates and other crystals.Metallurgy, Ceramics. FLUORESCENCE MICROSCOPY:- Principle: Fluorescence is the property of some substances which, when illuminated by light of a certain wavelength, will re-emit the light at a longer wavelength. In fluorescence microscopy, the exciting radiation is usually ultra violet wavelength or blue region. A substance which possesses a fluorophore will fluoresce naturally (Primary or autofluorescence) eg Vitamin A, chlorophyll Dyes, chemicals and antibodies added to tissues produce secondary fluorescence of structures and are called fluorochromes. When antibodies labeled with fluorochromes are used to detect particular antigens, the technique is called Immunofluorescent technique and is widely used in medicine. Tissue antigens most commonly demonstrable by Immunofluorescence are viruses, protozoa, bacteria, enzymes, hormones, plasma proteins, cells and cell constituents. Examples of fluorochromes: Fluorescein (apple green emission color), Rhodamine (Orange-red color

Light microscope
Introduction:- Light Microscope instruments are designed to produce magnified visual or photographic images of objects too small to be seen with the naked eye. The Light Microscope must accomplish three tasks: produce a magnified image of the specimen, separate the details in the image, and render the details visible to the human eye or camera. This group of instruments includes not only multiple-lens (compound Light Microscopes) designs with objectives and condensers, but also very simple single lens instruments that are often hand-held, such as a loupe or magnifying glass. Objectives of Light Microscope Study After reading this lesson, you will be able to: *describe the principle of light microscope *explain the parts of a light microscope *learn how to use a microscope.LIGHT AND ITS PROPERTIES:- Light radiates in all directions, with each ray traveling straight till infinity, unless Amplitude:- refers to the strength of energy or brightness of light. When light passes through any medium, the amplitude decreases depending upon the medium. Wavelength:- The distance between the apex of one wave and the next is the wavelength and measured in nanometers, and determines the color. Retardation:- Media through which light is able to pass, will slow down the speed of light (proportionate to density of medium). Refraction:- If light enters a medium (eg glass) at an angle, a deviation of direction occurs Image Formation . Focal point:- Parallel rays entering a simple lens are brought together to a single point called focal point, where a clear image will be formed. Conjugate foci:– Object placed at one end of lens will form a clear image on a screen kept at other side of lens. Conjugate foci vary in position. If object is nearer the lens, the image will be formed further away, at a greater magnification and inverted. This “real” image is formed by objective lens of microscope. If the object is placed within focal point of lens, image is formed on same side as object, is enlarged, right way up and cannot be projected on a screen, this is the “virtual image”. The eye piece of microscope forms this image Image Quality:- As white light is composed of all spectral colors, different wavelengths will be refracted to different extent. This lens defect is called chromatic aberration.Spherical aberration is caused when light rays entering at a periphery are refracted more than those entering the centre of lens. Both these faults can be corrected by using combination of lenses and lens elements. COMPONENTS OF A MICROSCOPE:- Light source:- Light source can be external or inbuilt. Dispersal of heat, collection of greatest amout of light, direction and distance are carefully calculated by the designers of microscope for greatest efficiency. Condensers:- The purpose of condenser is to concentrate the light into the plane of the object. The more the light at the specimen, better is its resolution. All condensers have aperture diaphragm with which the diameter of the light beam can be controlled. Object stage :- It is a rigid platform with an aperture through which the light can pass. It supports the glass slide. It allows controlled movement in two directions. Objectives :- They are the most important parts of microscope. The main task of objective is to collect the maximum amount of light from the object, unite it and form a high quality magnified real image. Magnifying powers of objectives are from 1:1 to 100:1. Body tube:- Body tube can be monocular, binocular and the combine photo-binocular (also called trinocular). Binocular tubes have provision for inter-pupillary distance adjustment, enabling each observer to adjust for his eyes. Eyepiece:- The final stage in optical path, the eyepiece’s function is to magnify the image formed by the objective within the body tube, and present the eye with a virtual image. Use of the Microscope:-
Introduction of histopathology
Introduction:- Surgical pathology includes gross and microscopic examination of resected specimens and biopsies by histopathologists for tissue diagnosis. Several steps are followed to get the tissue in a form, by which diagnosis can be made under light microscope Objective:- After reading this lesson, you will be able to: list the steps involved in the processing of surgical specimens for histopathologic examination explain the after care of the specimens explain grossing and gross room describe the laboratory hazards and safety measures. Steps involved in the process are 1. Receipt of specimens from OT 2. Grossing 3. Tissue processing 4. Embedding 5. Section cutting 6. Staining and labelling 7. Dispatch of slides to pathologist for diagnosis
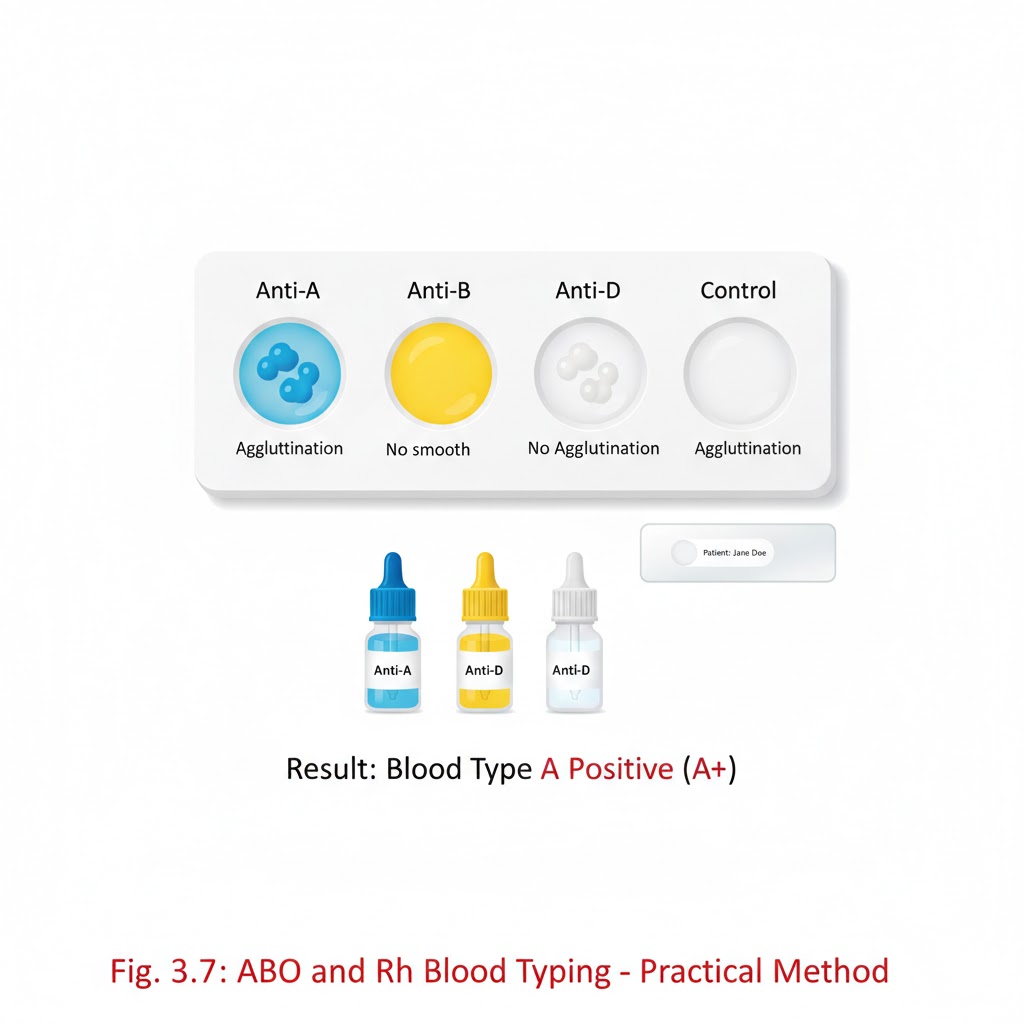
PRACTICAL OF ABO AND Rh TYPING
ABO AND Rh TYPING IS PERFORMED BY TWO METHOD:- 1.SLIDE METHOD:- Aim: – ABO grouping and Rh typing using the slide method. Objective: – ABO grouping and Rh typing using the slide method, which is a standard procedure for determining an individual’s ABO and Rh blood types. Principle: – ABO grouping and Rh typing performed by antisera based on the principle of agglutination. Normal human red cell processing antigen will clump in the presence of corresponding antibody (Anti A, Anti B, Anti D). Requirement:- *whole blood sample (EDTA) *Disposable gloves *Clean glass slide *ABO grouping and Rh typing reagents *Toothpicks or wooden sticks *Droppers of pipettes *Absorbent paper towels *Timer or stopwatch *Blood typing record sheet. Preparation:- Ensure that you are working in a well-lit and clean laboratory environment Label each glass slide with the patient’s identification or sample number Put on disposable gloves to maintain aseptic conditions ABO blood grouping procedure:-*Take three clean and dry glass slides *Place one drop of patient’s blood on each slide *Add a drop of Anti A reagent on first slide anti B on second slide, Anti D on third slide. *Mix the blood and serum gently using separate wooden sticks. Observation and record the reaction: – *if blood agglutinates (clumps)with anti A serum then the patient blood group is A. *If blood agglutinates with anti B serum, then the patient blood group is B. *If blood agglutinates with anti A and anti B both serums, then the blood group is AB. *If blood do not agglutinate with both anti A and anti B serum, then the blood group is O. Antisera A Antisera B Blood group + – A – + B + + AB – – O Rh blood grouping procedure: – Take clean and dry glass slide Place a fresh drop of the patient blood on slide Add a drop of anti Rh (anti D) serum. Mix gently with wooden stick. Observation and record the reactions: – if the blood agglutinates with anti Rh serum, then the patient blood Rh positive e.g. (A+, B+, AB+, O+) If blood is not agglutinate with anti Rh serum, then the patient blood Rh negative e.g. (A-, B-, AB-, O-) Antisera D Blood group + Rh positive – Rh negative Clinical significance: – ABO grouping and Rh typing is important in: – *.Blood transfusion *.Used for the personal identification and paternity exclusion (before DNA testing) *.Before a person donates blood. *.Before an organ and tissue transplant *.Before surgery *.To show whether to people could be blood relatives *.To check the identity of a person suspected of committing a crime. Note: – The ABO and Rh typing determination is used for the transfusion safety, pregnancy management, transplantation, forensic identification, and understanding disease susceptibility. TEST TUBE METHOD:- AIM: – ABO grouping and Rh typing by test tube method. Principle: –In the ABO grouping and Rh typing using the antisera is based on the principle of agglutination. Normal human red cell processing antigen will clump in the presence of corresponding antibody. Requirements: – blood group tube Pasteur pipettes. Centrifuge. Reagent. Normal saline. Blood sample. Procedure: – Prepare 5% red cell suspension for ABO grouping and Rh typing. Mix 5 drops(0.05ml) of sediment red cell with 2 ml of normal saline. Centrifuge at 1500 rpm for 1–2-minute discard the supernatant part wash 3 time with normal saline. Add 4ml of normal saline to sedimented red blood cells. Take 3 test tube label them as A, B, D. Place 1 drop of anti A into ‘A’ tube, one drop of anti B into the ‘B’ tube. one drop of anti D in ‘D’ tube. Add one drop of RBC suspension to each tube. Gently shake each tube to mix the contents. And then centrifuge tube at 1500 rpm for 1 minute. The RBC will form a button or pellet at the bottom of each test tube. Gently resuspend the RBC button and examine agglutination macro and microscopically. Observation: – Antisera A Antisera B Blood group + – A – + B + + AB – – O Antisera D Blood group + Rh positive – Rh negative
human blood group system
Human blood group system:- Rh are major, the most important of all the blood group system. All people (with some exception) of ABO system can be divided in 4 major groups in this system they are A, B, AB and O group. This determines by the reaction of two different reagent, known Anti A, and Anti B. For example: – Forword grouping or cell grouping by using known Anti A and Anti B results obtained are as follow. Red cell sample Reagent Reaction Blood group Red cell Anti A Agglutination A groupThere are nearly 300 blood group systems so for discovered. The ABO and Red cell Anti B No agglutination Red cell Anti A No agglutination B group Red cell Anti B Agglutination Red cell Anti A Agglutination AB group Red cell Anti B Agglutination Red cell Anti A No agglutination O group Red cell Anti B No agglutination ABO BLOOD GROUP SYSTEM:- Karl Landsteiner, an Austrian scientist discovered the ABO blood group system in the year 1900.in his experiments, he mixed different blood types and noted that the plasma from certain blood type produced agglutinates. Which were caused by the absence of molecules on red blood cells and resulting in antibodies to defeat that molecules. He made a note of the agglutination and divided the blood type in 4 different groups. For the discovery of ABO blood group, he was awarded the Nobel price. ABO and Rh blood group: – During blood transfusion, two group system are examine the ABO and Rh (Rhesus)system. ABO BLOOD GROUP: – The ABO system is based on the presence or absence of two antigens (A and B) on the surface of red blood cells (RBCs). The presence of A,B, or O antigen on red cell is determined by the inheritance of the allelic genes A, B, and O on chromosome 9, which are inherited in pairs as Mendelian dominants. it classified into 4 types *Group A → RBCs have A antigen, plasma has anti-B antibodies *Group B → RBCs have B antigen, plasma has anti-A antibodies *Group AB → RBCs have both A and B antigens, plasma has no antibodies (universal recipient) *Group O → RBCs have no antigens; plasma has both anti-A and anti-B antibody The ABO group system is important in blood transfusion during blood donation as mismatching of blood group can lead to clumping of RBCs with various disorders. It is important for blood cells matching during transfusing i.e. donor -recipient compatibility is necessary. ABO antibody: – ABO antibodies are usually IgM, which are cold reacting. These antibodies don’t cross the placenta and can bind complements. The majority of anti A from group B person or individual and anti B from group A person or individual contain IgM antibody with minor amount of IgG or IgA present. Antisera used in ABO test procedure: – In ABO test procedure antigens present on an individual cells are determining method is called forward typing or direct typing and consist of testing the unknown cells with known antisera and then observing for agglutination. Anti A antiserum: – The serum is obtained from “B” type individuals, since there is a “natural” occurrence of anti A agglutinin in their serum. Anti B antiserum: – The serum is obtained from “A” type individuals where there is a natural occurrence of anti B agglutinin in the serum. Anti A, B typing serum: – This anti serum is obtained from “O” Type individuals. Anti A1: – This serum is obtained from human sources or plant lectins. This serum determines A1 and A1 B person from other A and AB types. Anti H: – this serum is obtained from lectin (plant source) this antiserum determines Bombay (oh)types (who lack H substance 2. Rh (Rhesus) system: – The Rh system is the second most important blood group system after the ABO system in transfusion medicine. About two-third of the papulation contain the third antigen on the surface of their red blood cells known as Rh factor or Rh antigen. It was first discovered in 1940 by Landsteiner and Wiener in his experiment using the blood of Rhesus monkeys NOTE: – The Rh system is a complex blood group system, mainly determined by the D antigen, playing a vital role in transfusion safety and in preventing haemolytic disease of the newborn The Rh system has more than 49 antigens, but the most significant is the D antigen. If a person has the D antigen on their red blood cell, then the person is Rh positive (Rh+). If D antigen absent on their red blood cell, then the person is Rh negative (Rh-) Some other important Rh antigens are C, c, E, and e. The Rh system is inherited genetically. It is controlled by two closely linked genes on chromosome 1. RHD gene – produces the D antigen. RHCE gene – produces C/c and E/e antigen. Haemolytic disease (break down of RBC called haemolytic disease) of the newborn (HDN):-occurs when an Rh- mother carries an Rh+ foetus. the mother’s immune system may produce anti D antibodies that destroyed fatal red blood cells. Blood Transfusion Reactions:If Rh⁻ patients receive Rh⁺ blood, they may form antibodies (anti-D), leading to haemolysis in future transfusions. That’s why Rh typing and cross-matching are essential before blood transfusion. Rh⁻ mothers are given anti-D immunoglobulin after delivery or miscarriage to prevent sensitization. Note: – Bombay blood group is discovered by Y.M Bende in 1950.

CROSS MATCH
CROSS MATCH Cross matching is a laboratory test that checks the compatibility between the donor’s red blood cells (RBCs) and the recipient’s serum(plasma). it helps to prevent transfusion reactions caused by antibodies in the recipient attacking donor red blood cells. or cross match is the final compatibility test is performed in a blood bank to ensure that a patient blood is compatible with a donor blood before transfusion. If blood is compatible with recipient blood after cross matching, then donor can donate blood. if not compatible then donor cannot donate blood. The cross match is a final check reverse and forward. In 1907 first time Hekaton he just proposes cross match. In 1908 Ottenberg first time use cross match for blood transfusion in New York. Procedure: – *Make 5% cell suspension for both donors and recipients. *Take two clean and dry test tubes and mark as major and minor cross match. *Add 50µl donor cell suspension (antigen)and 25µl serum (antibody)of recipient for major cross match. *Add 50µl cell suspension of recipient and 25µl serum of donor for minor cross match. *Mix well and incubate for 30 minutes at 37°c *After incubation observe microscopically and macroscopically Observation: – If agglutination shows, then blood is not compatible for recipient. If agglutination is not shown, then the blood is compatible for recipient. Cross match by micro gel tube method: – The micro gel tube method, also called the gel card method, is a modern and reliable technique used in blood bank to perform cross matching between donor and recipient blood. It helps ensure that compatibility before transfusion, preventing transfusion reactions. Principle: – The gel microcolumn contains antiglobulin reagent (coombs reagent) or other media that trap agglutinated red cells during centrifugation. Agglutinated(incompatible)red cells are trapped in the gel. Non agglutinated(compatible) red cells pass through the gel and settle at the bottom. It detects antigen-antibody reactions between donor red cells and recipient serum/plasma. Requirement: – Gel card (antiglobulin gel microtube card) Sample (both donor and recipient) Micropipette and tips Centrifuge (specific for gel cards) Incubator Procedure: – Prepare 2-5% suspension of donor red cells in normal saline. Add 50µl of recipient serum/plasma into the designated microtube of gel card. Add 25µl of donor red cell suspension to the same microtube. Incubate the card at 37°c for 15-30 minutes (to allow antigen-antibody reaction). Centrifuge the card in a gel card centrifuge for 10 minutes. Observe the position of red cells in the gel column. Interpretation or Results: – If red cells are forming a band at the top or dispersed in the gel then result positive means agglutination occurred, blood is incompatible. If red cells are forming a pellet at the bottom of the microtube (or RBC settle down in bottom of microtube) then result negative means agglutination does not occur, blood is compatible. If mixed pattern means some cells are trapped, or on top and some at bottom then result weak positive show incompatibility (possible minor).