Cerebrospinal fluid (CSF) examination
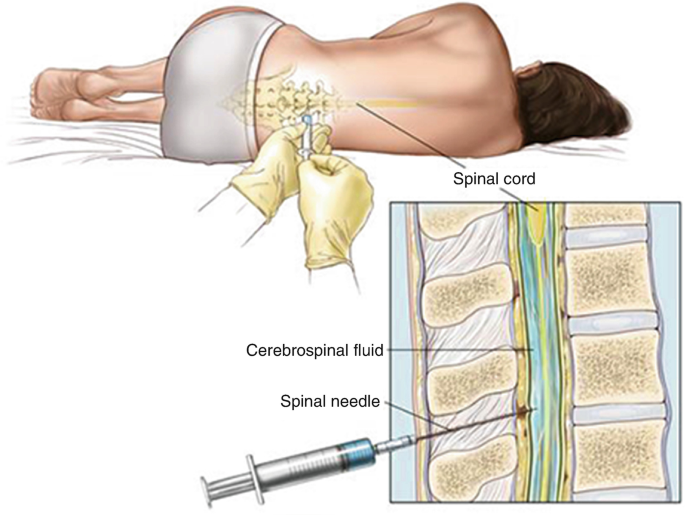
Cerebrospinal fluid(CSF):- CSF are produced by the choroid plexus of the lateral, third, and fourth ventricles up to 80% approximetly and 20% CSF are produced by the surface of brain and spinal card. CSF is a clear, colorless, sterile fluid that fills the ventricles of the brain, the central canal of the spinal cord, and the subarachnoid space surrounding the brain and spinal cord. CSF produced about 500 mL per day, although only about 150 mL is present at any given time because it is continuously reabsorbed into the bloodstream through the arachnoid villi and granulations. CSF circulates through the ventricular system and around the central nervous system, forming a protective fluid environment for neural tissues. CSF performs several essential physiological functions. It acts as a mechanical cushion, shocks absorbing and protecting the delicate brain and spinal cord from trauma. It provides buoyancy, reducing the effective weight of the brain.CSF also helps maintain chemical homeostasis The CSF examination or dignosis is important for diagnose infections such as Meningitis and Encephalitis, inflammatory disorders such as Multiple Sclerosis, hemorrhages, malignancies, and other neurological conditions. CSF acts as a mechanical cushion for the brain and spinal cord, absorbing shocks and protecting them from injury caused by sudden movements or impacts. CSF collection :- SF collection is the procedure of obtaining a sample of cerebrospinal fluid (CSF) for laboratory examination to diagnose diseases.The most common method of CSF collection is lumbar puncture (spinal tap).The lumber puncture is done by expert physician , surgeon & Nurses. Procedure for CSF collection:- 1.The patient is usually placed in a lateral decubitus (side-lying) or sitting position. 2.Cleaning the skin and administering local anesthesia. 3.A sterile spinal needle is inserted into the subarachnoid space between the L3–L4 or L4–L5 vertebrae,below the end of the spinal cord. 4.The needle enters the subarachnoid space, CSF flows through the needle and is collected into sterile tubes. 5.Specimen centrifuged. then the supernatant part used for biochemical tests and the sediment is used for weight mount preparation ,gram’s staining , etc. Physical examination of CSF:– It consist of following examination:- 1.Odour:- CSF are odourless normally in few of bacterial infection conditions putrified small occures. 2. Colour:- Colourless but in bacterial menengitis & physical damage causes redish colour CSF. 3.pH:- Normally pH of CSF is 7.35 – 7.4. 4.Volume:- In adults 120-150 ml CSF are flow in around the spinal card & brain,In child 80-120 ml CSF and neonate 10-60 ml.The produced rate of CSF is 500 ml per day. 5.Specific gravity:- Normal specific gravity 1.003-1.008. 6.Appearence:- Normally CSF are clear but in bacterial menengitis. its may cloudly or Frankly purulent.(Frankly- clear or clean, purulent-containing pus.). Chemical examination:- CSF Sugar GOD/POD Methods:- Aim:– Performed CSF sugar test by GOD/POD (Glucose oxidase & peroxydase) method. Principle:- 1. Glucose + H₂O Glucose Oxidase Gluconic acid +H₂O₂ 2. H₂O₂ + 4AAP(Aminoantipyrine) + Phenol Peroxidase Quinoneimine dye. 1. Take a three clean and dry test tube and mark as blank standerd and test. 2.Measures all contents according to chart. 3.Mixwell and incubate for 10 minutes at 37 degree celcious temperatures. 4. Read the optical density of the test and standered against blank.at500-520 nm wave length. Calculation:- CSF glucose calculation = OD of test × Concentration of Standard OD of standered Concentration of Standard = 100 mg/dl Result:-…………….? Normal Value:- 60-80 mg/dl Clinical significance:- Decreased CSF Glucose (Hypoglycorrhachia):- Microorganisms and inflammatory cells consume glucose, lowering its concentration in CSF in condition Bacterial meningitis,tuberculous meningitis, fungal meningitis , malignant infiltration of the meninges,severe CNS inflammation. Increased CSF glucose(hyperglycemia):- CSF glucose generally rises in proportion to blood glucose levels.hyperglycemia (Diabetes mellitus). -:CSF Microprotein:- Aim:- Performed CSF protein by biurate methods. Principle:- CSF protein + Cu2 Alkaline medium Violet color complex (Cu-Protein Complex) Requirment:- Test tube, micro-pepette,D/W , calorimeter, CSF sample, working reagen , incubator, tissue paper. 1. Take three clean and dry test tube and mark as blank standerd and test. 2.Measures all contents according to chart. 3.Mixwell and incubate for 5 minutes at 37 degree celcious temperatures. 4. After incbation measure and read the optical density of the test and standered against blank at 540 nm wave length. Calculation:- CSF protein = OD of test × Concentration of Standard OD of standered Concentration of Standard = 6 mg/dl Result:-.………………………? Normal value:- 15-45 mg/dl Clinical significance:- CSF microprotein estimation helps diagnose neurological disorders. Increased CSF protein is observed in meningitis, tuberculous