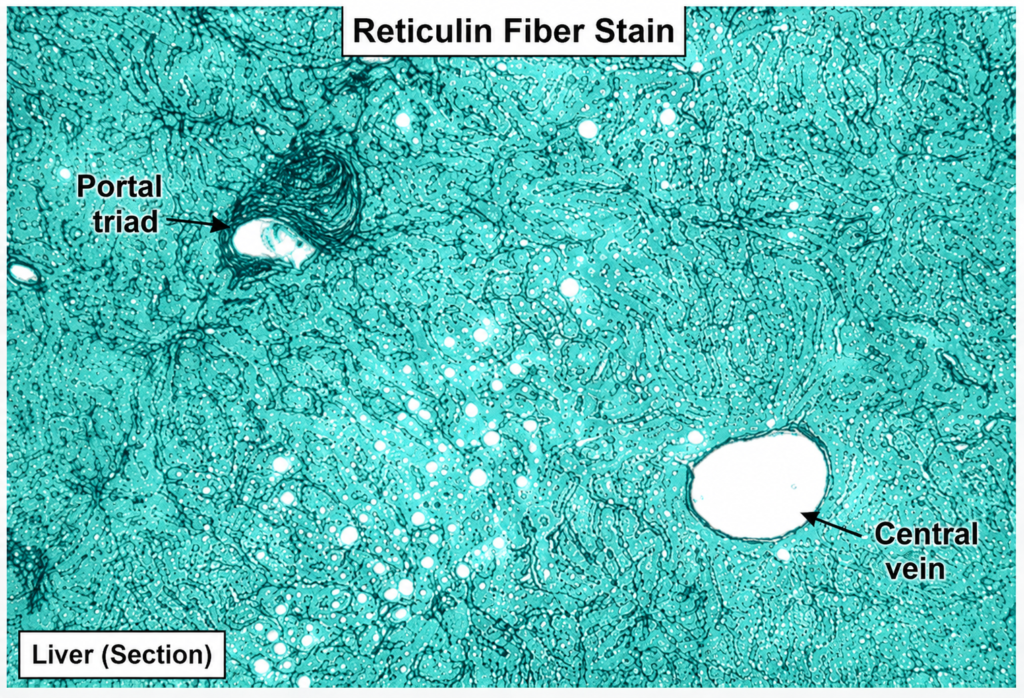
HORMONAL ASSESSMENT REPORTS
INTRODUCTION:- The established approach to the evaluation of ovarian function and endocrine disorders in the woman is based on serial biochemical analyses of hormones, such as estrogen, progesterone, luteinizing hormones and their metabolites. In women who suffer from menstrual disorders and abnormalities of the ovarian cycle, the biochemical analyses can be effectively supplemented by the old fashioned endometrial biopsies, or studies of endocervical mucus. In addition, the cervicovaginal smear may sometimes provide useful information and has the advantage of being easy to obtain, rapidly evaluated, and inexpensive. The cytologic approach is particularly valuable if laboratories specializing in endocrine analysis are not readily available. The principle of the cytologic hormonal analysis is simple. The degree of maturation of the squamous epithelium of the female genital tract depends on steroid hormones, mainly estrogen. HORMONAL ASSESSMENT:- Naturally occurring estrogen, or the parenteral administration of estrogen or its natural or synthetic substitutes in adequate amounts, produces a rapid and complete maturation of the normal squamous epithelium of the female genital tract with a resulting preponderance of mature superficial squamous cells in smears. The effect takes place regardless of the prior hormonal status, except during pregnancy. Conversely, complete atrophy of the squamous epithelium of the vagina and cervix may be equated with complete absence of estrogenic activity. However, there are no reliable data linking intermediate degrees of maturation of the squamous epithelium with the action of a specific hormone or hormones. Evaluation of the endocrine status of a menstruating woman during the childbearing age belongs among the most difficult tasks in diagnostic cytology. There is considerable variation in the smear patterns from one patient to another, even if matched for age and menstrual history. Several conditions must be fulfilled before a successful hormonal evaluation of the squamous epithelium may be undertaken. 1.There must be absence of inflammation or cytolysis. 2.There must be no recent medication, either topical or systemic, especially with compounds known to affect the squamous epithelium of the lower genital tract. 3.There must be no history of radiotherapy or recent surgery to the vagina or cervix. 4.An adequate baseline investigation must have been performed in menstruating women. This should include daily smears during at least one and preferably two complete cycles, or their chronologic equivalent. In nonmenstruating patients, two or three smears may suffice. 5.The smears should be obtained from the proximal portion of the lateral wall of the vagina, care being taken to avoid contamination with material from the adjacent cervix. The Karyopyknotic Index (KI):- The karyopyknotic index expresses the percentile relationship of superficial squamous cells with pyknotic nuclei to all mature squamous cells. Usually, 200 to 400 consecutive cells in three or four different fields on the smear areevaluated. The peak of KI usually coincides with the time of ovulation and was estimated at 50% to 85% of total cells. The Eosinophilic Index (EI):- The eosinophilic index expresses the percentile relationship of mature squamous cells with eosinophilic cytoplasm to all mature squamous cells, regardless of the status of the nucleus. In a normal menstruating woman, the peak of EI coincideswith the peak of KI and may reach 50% to 75% at the time of ovulation. The Maturation Index (MI):- The maturation index expresses the maturation of the squamous epithelium as a percentile relationship of parabasal cells to intermediate cells to superficial cells. The count should be performed on single cells. For example, in a normalmenstruating woman at the time of ovulation, an MI of 0:35:65 would indicate that the smear contained no parabasal cells, 35% of intermediate cells, and 65% of superficial cells. Other Indices:- The folded-cell index represents the relationship of mature superficial or intermediate squamous cells with folded cytoplasm to all mature squamous cells. The crowded-cell index represents the relationship of mature squamouscells lying in clusters of four or more cells to all mature squamous cells. Alternative Ways of Reporting Hormonal Status:- It has been a common practice to base the evaluation of the maturation of the squamous epithelium on an overall visual impression gained during the routine screening of smears. This simplest of methods has not failed in revealing majorabnormalities of smear patterns. By comparing the current smear pattern with original baseline smears, a good appreciation of changes in smear pattern may be gained. Small variations in smear pattern have no diagnostic meaning but maystrongly influence the indices and thus give a false impression of hormonal “effects.” The reporting of smears based on this overall visual impression is always given in reference to age, menstrual history, and possible clinical significance. Some examples follow: Patient age 35: “Midcycle smear pattern—consistent with functioning ovaries.” Patient age 52: “Absence of maturation of squamous cells consistent withmenopause.” Patient age 25: “Absence of maturation of squamous cells—abnormal for age.” Patient age 60: “High level of maturation of squamous cells not consistent with clinical menopause. It is assumed that this patient is not receiving estrogens or other drugs that may account for this smear pattern.” DETERMINATION OF THE TIME OF OVULATION FROM CERVICOVAGINAL SMEARS:- A precise determination of the time of the ovulation is important in artificial insemination and in in-vitro fertilization. The use of the cervicovaginal smears to establish the time of ovulation or the status of the endometrium has been of limited reliability. It is recommended that cytologic methods for estimation of ovulation or status of the endometrium be supplemented by other procedures, such as temperature curves and endometrial biopsies. The examination of endocervical mucus may also be of assistance. Cyclic changes in the physicochemical properties of the cervical mucus have been known for a great many years. Prior to ovulation, the mucus tends to be viscous and when placed on a glass slide, form crystalline, fern-like structures, whereas at the time of and after ovulation, the mucus is more liquid and does not crystallize. Cytologic evaluation for menstrual abnormalities:- 1. Cytologic hormonal evaluation may be of assistance in the evaluation of amenorrhea (cessation of menses), in women who have never menstruated (primary amenorrhea) or who stopped menstruating at a young age after