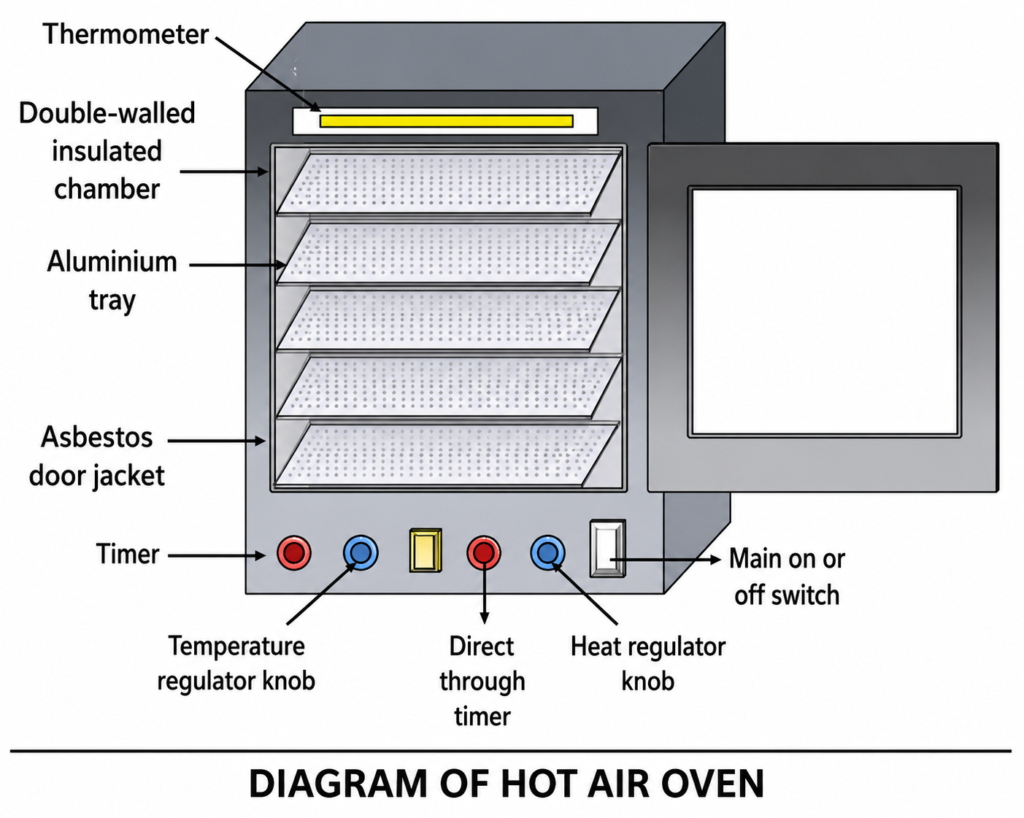
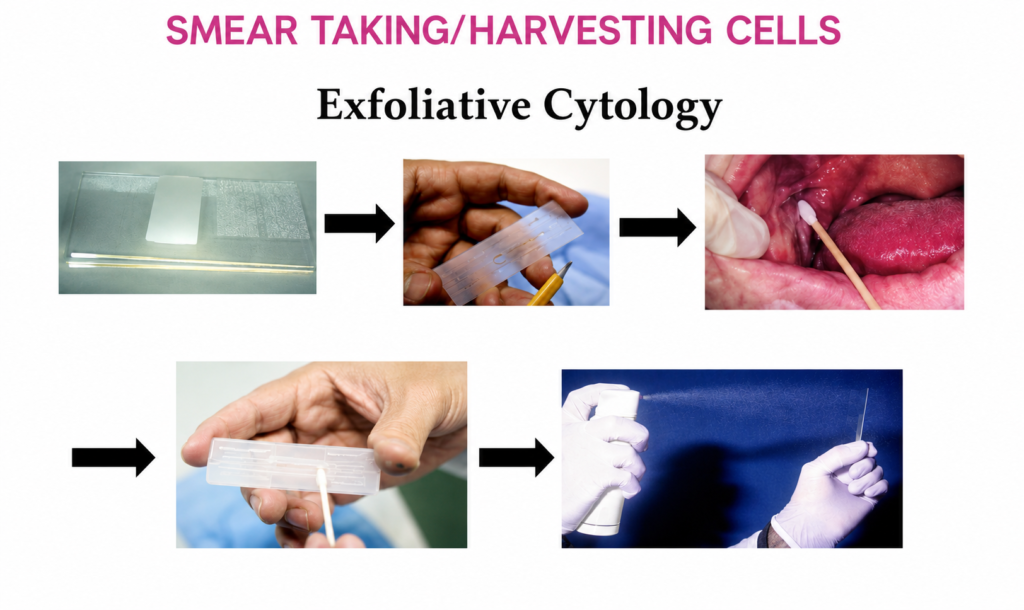
INTRODUCTION:- Exfoliative cytology, which is a quick and simple procedure, is an important alternative to biopsy in certain situations. In exfoliative cytology, cells shed from body surfaces, such as the inside of the mouth, are collected and examined. This technique is useful only for the examination of surface cells and often requires additional cytological analysis to confirm the results. Exfoliative cytology differs from the more precise sampling of known lesions, like needle biopsy. It categorizes collected samples only by analyzing the presence of abnormal or atypical cells, or by showing the presence of malignant cells. When a woman has a pap smear, she may have a result that show atypical cells. If this is the first exfoliative cytology test that shows atypical cells, then usually, the Pap smear is repeated in six to twelve months. If however, repeated showings of atypical cells are present in exfoliative cytology results, further tests may be undertaken to determine if cancerous cells are present. Doctors or dentists may also use exfoliative cytology to check for the presence of cancer in the mouth or throat. The test takes a few skin scrapings and can show the presence of either malignant or atypical cells. Malignant and atypical cells will probably require a person to undergo a biopsy or closer examination of the area in question to rule out cancer. Cytologic examination of a serous effusion is of paramount importance because the finding of cancer cells in such a specimen denotes that the patient has cancer that is not only advanced but also almost always incurable. Apart from the finding of cancer cells, cytologic examination of pleural, peritoneal, and pericardial effusions may also reveal information about inflammatory conditions of the serous membranes, parasitic infestations, and infection with bacteria, fungi, or viruses. COLLECTION METHOD:- In this method, cells are collected after they have been either spontaneously shed by the body (“spontaneous exfoliation”) or manually scraped/brushed off of a surface in the body (“mechanical exfoliation”). An example of spontaneous exfoliation is when cells of the pleural cavity or peritoneal cavity are shed into the pleural or peritoneal fluid. This fluid can be collected via various methods for examination. Examples of mechanical exfoliation include Pap smears, where cells are scraped from the cervix with a cervical spatula, or bronchial brushings, where a bronchoscope is inserted into the trachea and used to evaluate a visible lesion by brushing cells from its surface and subjecting them to cytopathologic analysis. Spontaneous exfoliation: Peritoneal fluid, pleural fluid, pericardial fluid, urine, cysts, washings (peritoneal, bladder) The fluid is collected into a clean, dry container, which need not be sterile, and sent to the laboratory as soon as possible. If the fluid cannot be sent immediately, it should be stored in a refrigerator at 40C and not allowed to freeze. We do not require anticoagulant or fixative to be added to the fluid. The appearance to the naked eye of a serous effusion sometimes reveals clues about the cause of the effusion and the nature of its cellular contents. Therefore, for every serous effusion received by the laboratory, note should be made of its volume, color, clarity, and any unusual physical features, such as malodor, opalescence, or high viscosity Mechanical exfoliation: Cervical pap smear, brushings (Bronchial, gastric, biliary, oral, etc). Cervical smear is a reliable method for diagnosis of cervical cancer. The smears are usually taken in Gynecology ward or OPD, but sometimes patients are sent to laboratory for smear purposes. Patients should be advised NOT to douche, use vaginal medications, or have intercourse 24 hours prior to the pap smear preparation. Patients should NOT schedule pap smear exams during menses. These situations may obscure cellular details or remove diagnostic material from the cervix or vagina. The smear is obtained under direct vision after introduction of speculum. A wooden tongue depressor cut with scissors to fit the contour of cervix may be used. Commercially prepared plastic or wooden scrapers are widely available for this purpose. The scraper is rotated under pressure to 360° for 4-5 full rotations. The material is spread on a pre labelled slide and fixed immediately. Several types of brushes have also been introduced to overcome the disadvantages of scrapers alone (not being able to reach endocervical canal and transformation zone where the carcinomas originate). Brushes are also used to scrape cells in respiratory tract, oral mucosa, esophagus, stomach, duodenum, colon and biliary tract. It is preferable to obtain the brush sample before the biopsy because the latter results in bleeding, which both obscures the lesion, and detracts from the quality of a subsequently collected cytologic sample, whereas interpretation of the biopsy is not affected by the reverse order of collection. Collection of a good brush sample usually requires an experienced assistant, because the operator may well be engaged in maneuvering the end of the scope and holding the lesion in focus while the assistant manipulates the brush. Therefore, it is ideal that a Cytology staff member be present for immediate slide preparation of the specimen. They are all taken under vision- direct or through fibreoptic endoscopy. In all these cases a lot of care needs to be taken to make smear immediately from the brush by gently rotating the brush on slides and fixing them immediately. The material should not be crushed. If liquid-based cytology is used, the head of the broom is detached and dropped into the preservative vial. Once received in laboratory, the usual precautions need to be taken as discussed in specimen receiving, handling and storage. When only one slide is received, it should be preferable to stain it with Papanicolaou stain. When multiple slides are received- some are air dried (stain with MGG, special stains, etc) and some are wet fixed (stain with Papanicolaou). The liquid specimens need to be commented upon the volume, color and turbidity. Specimens need to be centrifuged and cytocentrifuged depending on cellularity. After concentrating they can be used for both air dried smears and wet fixation.