Cavities of the body
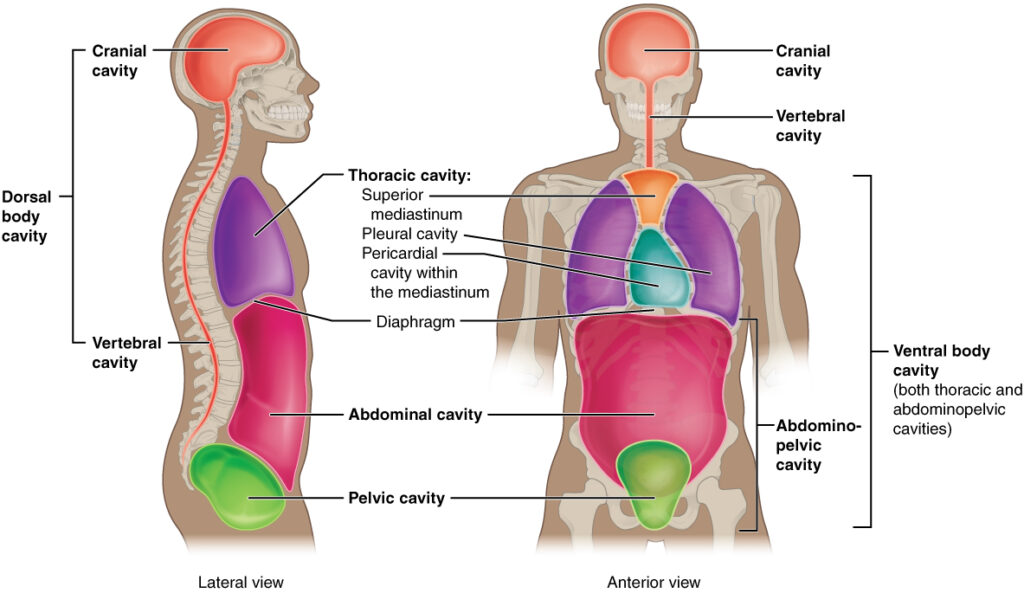
The human body cavity is a fluid-filled space inside the body that holds and protects internal organs. The body maintains its internal organization by membrane,sheaths,and other structure that separate the compartments. The two main divisions are the dorsal cavity (posterior) and ventral cavity (anterior).These cavities protect the lungs,heart,stomach,and intestines for example can expand and contract without distorting other tissues or disrupting the activity of nearby organs. The posterior (dorsal) cavity has two main subdivisions:- 1.cranial cavity :- The cranial cavity is the hollow space inside the skull and protects the brain.The cranial cavity contains the brain,protective membranes called meninges. cerebrospinal fluid (CSF) blood vessels supplying the brain.its called as houses of the brain.its formed by the bones of the skull especially the cranium protected by skull and cerebrospinal fluid. Functions of the Cranial Cavity:- Brain Protection: The solid, bony walls (calvarium) and fluid-filled membranes act as a shock-absorbing enclosure for the brain, cerebellum, and brainstem. Structural Support: The base of the skull forms the floor, consisting of the anterior, middle, and posterior cranial fossae, which provide a stable, shaped foundation where different parts of the brain rest. Muscular Attachment: The exterior surface of the cavity provides anchoring points for muscles, including the temporalis muscle, which is vital for jaw movement. Environmental Stability: It creates a stable, insulated environment necessary for delicate brain functions, including the regulation of temperature, hormonal signals via the pituitary gland, and blood flow. vertibral cavity:-The spinal cavity (vertebral cavity) encloses the spinal cord .its a long, narrow space inside the vertebral column (backbone) Protected by the vertebral column and cerebrospinal fluid.It extends from the base of the skull to the lower back.the vertibral cavity main function is protect the spinal cord from injury and provides a pathway for spinal nerves.its supports communication between the brain and the rest of the body through the nervous system. Functions of the Vertebral Cavity:- Protection: The primary function is to serve as a strong bony encasement for the delicate spinal cord, protecting it from injury. Support & Structure: It forms a rigid central axis for the trunk, providing a safe housing for nerve tissue while supporting the body’s upright posture. Path for Nerves: The cavity enables nerve roots to pass out through the intervertebral foramina, facilitating connection to the peripheral nervous system. Space and Cushioning: It provides a contained space for the spinal cord, which is padded and held in place by cerebrospinal fluid and the meninges. Accommodation of Structure: The cavity allows for flexibility and movement of the vertebral column while maintaining the structural integrity of the spinal cord. The anterior (ventral) cavity has two main subdivisions:- 1.Thoracic cavity:-The thoracic cavity is situated between the neck and diaphragm in the upper part of the trunk.its boundaries are formed by the thoracic cage and supporting muscles.contain vital organs involved in respiration and circulation.(like-Trachea,2 bronchi,2 lungs,Heart, aorta, superior and inferior venacavae,blood vessels, oesophagus. The thoracic cavity is divided into three main compartments:- A. Right Pleural Cavity:- It contain the right lungs lined by pleura. B. Left Pleural Cavity :- It contain the left lungs also lined by pleura. C. Mediastinum (central compartment):-The Mediastinum is space between the lungs including the structures found there,sach as- heart oesophagus, and blood vessels Function of Thoracic cavity:- Protection: The rib cage, sternum, and thoracic vertebrae form a rigid, bony structure that shields vital organs such as the heart and lungs from injury. Respiration Facilitation: The cavity is designed for breathing (pulmonary ventilation). The diaphragm and intercostal muscles work to change the volume of the thoracic cavity, creating pressure changes that move air into and out of the lungs. Support & Movement: The cavity provides a stable anchor for the arms and protects the superior thoracic aperture (neck area), while remaining flexible enough for the torso to bend and twist. Organ Housing: It contains key cardiovascular components (heart, great vessels), respiratory components (lungs, trachea, bronchi), and the esophagus 2. Abdominopelvic cavities:-The abdominopelvic cavity is a large body cavity located below the diaphragm and above the pelvic floor is commonly divided into two parts: Abdominal cavity – the upper portion Pelvic cavity – the lower portion A.Abdominal cavity–The abdominal cavity is the upper part of the abdominopelvic cavity. The abdominal cavity is a large,fluid-lined body space that houses and protects the vital organs like-digestive,urinary,and reproductive organs. its the large hollow space in the body located between the chest(Thoracic cavity) and the pelvis. By convention,the abdominal cavity is divided into the nine regions:- 1.Epigastric region 2.Umbilical region 3.Hypogastric region 4. Left hypochondriac region